CIF Parser¶
This section covers working with CIF files on a syntactic level.
Higher-level functions that understand semantics of different types of CIF files are documented in other sections. These types include:
small molecule or inorganic CIF files,
macromolecular PDBx/mmCIF with coordinates,
PDBx/mmCIF with reflection data,
monomer/ligand files used to supplement macromolecular coordinates.
What are STAR, CIF, DDL, mmCIF?¶
(in case someone comes here looking for a serialization format)
STAR is a human-readable data serialization format (think XML or JSON) that happens to be known and used only in molecular-structure sciences.
CIF (Crystallographic Information File) – a file format used in crystallography – is a restricted derivative of STAR. It is restricted in features (to make implementation easier) but also imposes arbitrary limits – for example, on line length.
DDL is a schema language for STAR/CIF.
All of them (STAR, CIF and DDL) have multiple versions. We will be more specific in the following sections.
The STAR/CIF syntax is relatively simple but may be confusing at first. (Note that the initial version of STAR was published by Sydney Hall in 1991 – before XML and long before JSON and YAML, not to mention TOML.)
Key-value pairs have the form:
_year 2017 # means year = 2017
Blocks (sections) begin with the data_ keyword
with a block name glued to it:
data_tomato # [tomato]
_color red # color = "red"
Importantly, unlike XML/JSON/YAML/TOML, STAR is designed to concisely represent tabular data. This example from TOML docs:
# TOML
points = [ { x = 1, y = 2, z = 3 },
{ x = 7, y = 8, z = 9 },
{ x = 2, y = 4, z = 8 } ]
could be expressed as a so-called loop in STAR (keyword loop_
followed by column names followed by values):
# STAR/CIF
loop_
_points.x _points.y _points.z
1 2 3
7 8 9
2 4 8
Typically, long tables (loops) make up most of the CIF content:
1 N N . LEU A 11 ? 0.5281 0.5618 0.5305 -0.0327 -0.0621 0.0104
2 C CA . LEU A 11 ? 0.5446 0.5722 0.5396 -0.0317 -0.0632 0.0080
# many, many lines...
5331 S SD . MET C 238 ? 2.2952 2.3511 2.3275 -0.0895 0.0372 -0.0230
5332 C CE . MET C 238 ? 1.5699 1.6247 1.6108 -0.0907 0.0388 -0.0244
The dot and question mark in the example above are two null types.
In the CIF spec: ? = unknown and . = not applicable.
In mmCIF files . is used for mandatory items, ? for non-mandatory.
The CIF syntax has a flaw resulting from historical trade-offs:
strings interpretable as numbers do not need to be quoted.
Therefore, while the CIF specification defines a formal grammar for
numbers, we can’t recognize them. The type of 5332 is not certain:
the JSON equivalent could be either 5332 or "5332".
Note: “STAR File” used to be trademarked by IUCr and patented, but not anymore.
The mmCIF format (by mmCIF we mean what is more formally called PDBx/mmCIF) is the CIF syntax + a huge dictionary (ontology/schema) in DDL2. The dictionary defines relations between columns in different tables, which makes it resemble a relational database (it was designed at the height of popularity of RDBMSs).
Where are the specs?
International Tables for Crystallography Vol. G (2006) describes STAR, CIF 1.1, DDL1 and DDL2. If you don’t have access to it – the IUCr website has specs of CIF1.1 and DDLs. As far as I can tell all versions of the STAR spec are behind paywalls.
Later versions of the formats (hardly used as of 2017) are described in these articles: STAR (2012), DDLm and dREL (2012), and CIF 2.0 (2016). Only the last one is freely available.
PDBx/mmCIF is documented at mmcif.pdb.org. (More about it later.)
What is parsed?¶
The parser supports the CIF 1.1 spec and some extras.
Currently, it is available as:
C++14 header-only library, and
Python (3.8+) extension module.
We use it to read:
mmCIF files (both coordinates and structure factors)
CIF files from the Crystallography Open Database (COD)
Chemical Component Dictionary from PDB
DDL1 and DDL2 dictionaries from IUCr and PDB
monomer library a.k.a. Refmac dictionary
The parser handles:
all constructs of CIF 1.1 (including save frames),
the
global_andstop_keywords from STAR – needed for the Refmac monomer library andmmcif_nmr-star.dic, respectively.
It could be extended to handle the new features of CIF 2.0 or even the full STAR format, but we don’t have a good reason to do this. The same goes for DDLm/dREL.
The parser does not handle CIF 1.1 conventions, as they are not part
of the syntax:
line wrapping (eol;\eol),
Greek letters (\m → µ),
accented letters (\'o → ó),
special alphabetic characters (\%A → Å)
and other codes (\\infty → ∞).
CIF parsers in the small-molecules field need to deal with incorrect syntax. The papers about iotbx.cif and COD::CIF::Parser enumerate 12 and 10 common error categories, respectively. Nowadays the problem is less severe, especially in the MX community that embraced the CIF format later. So we’ve decided to add only the following rules to relax the syntax:
always
names and lines can have any length like in STAR (the CIF spec imposes a limit of 2048 characters, but some mmCIF files from PDB exceed it, e.g. 3j3q.cif),
quoted strings may contain non-ASCII characters (if nothing has changed one entry in the PDB has byte A0 corresponding to non-breaking space),
unquoted strings cannot start with keywords (STAR spec is ambiguous about this – see StarTools doc for details; this rule is actually about simplifying, not relaxing),
by default (set check_level=2 to throw errors in these cases)
a table (loop) can have no values if followed by a keyword or EOF (such files were written by old versions of Refmac and SHELXL, and were also present in the CCP4 monomer library),
block name (blockcode) can be empty, i.e. the block can start with bare
data_keyword (RELION and Buccaneer write such files),
as opt-in (set check_level=0 to tolerate these cases)
missing value in a key-value pair is allowed if whitespace after the tag ends with a new-line character,
duplicated block names, tags, and save frames are allowed.
DOM and SAX¶
The terms DOM and SAX originate from XML parsing, but they also became names of general parsing styles. Gemmi can parse CIF files in two ways that correspond to DOM and SAX:
The usual way is to parse a file or a string into a document (DOM) that can be easily accessed and manipulated.
Alternatively, from C++ only, one can define own PEGTL Actions for the grammar rules from
cif.hpp. These actions will be triggered while reading a CIF file.
This documentation covers the DOM parsing only. The hierarchy in the DOM reflects the structure of CIF 1.1:
Document contains blocks.
Block can contain name-value pairs, loops and frames.
Frame can contain name-value pairs and loops.
Loop (m×n table) contains n column names and m×n values.
Names are often called tags. The leading _ is usually treated
as part of the tag, not just a syntactic feature. So we store tag strings
with the underscore (_my_tag), and functions that take tags as arguments
expect strings starting with _.
The case of tags is preserved.
At this point it might help to show an example of DOM parsing.
Here is a simple program that reads an mmCIF file and prints
weights of components that are listed as _chem_comp.formula_weight.
The functions used here are documented later on.
// Compile with: c++ -I/path/to/gemmi/include -O2 my_program.cpp
#include <iostream>
#include <gemmi/cif.hpp>
namespace cif = gemmi::cif;
int main() {
try {
cif::Document doc = cif::read_file("1mru.cif");
for (cif::Block& block : doc.blocks)
for (auto cc : block.find("_chem_comp.", {"id", "formula_weight"}))
std::cout << cc[0] << " weights " << cc[1] << std::endl;
} catch (std::exception& e) {
std::cerr << "Oops. " << e.what() << std::endl;
}
}
import sys
from gemmi import cif
for path in sys.argv[1:]:
try:
doc = cif.read_file(path) # copy all the data from mmCIF file
for block in doc: # iterate over blocks
for cc in block.find("_chem_comp.", ["id", "formula_weight"]):
print(cc[0], "weights", cc[1])
except Exception as e:
print("Oops. %s" % e)
Values have types. CIF 1.1 defines four base types:
char (string)
uchar (case-insensitive string)
numb (number that cannot be recognized as number on the syntax level)
null (one of two possible nulls:
?and.)
Additionally, DDL2 dictionaries specify subtypes.
For example, int and float are mmCIF subtypes of numb.
Since in general it is not possible to infer type without the corresponding dictionary, the DOM stores raw strings (including quotes). They can later be converted to the required type using the following helper functions:
as_string()– gets unquoted stringas_number()– gets floating-point numberas_int()– gets integeras_char()– gets single characteris_null()– check if the value is null (i.e.?or.)
and we also have:
quote()– the opposite ofas_string()– adds quotes appropriate for the content of the string (usually, quotes are not necessary and not added).
#include <gemmi/cifdoc.hpp> // for as_string, as_int, quote
#include <gemmi/numb.hpp> // for as_number
double rfree = cif::as_number(raw_rfree_string); // NaN if it's '?' or '.'
cif::as_int("123"); // 123
cif::quote("two words"); // "'two words'"
cif::is_null("?"); // true
>>> from gemmi import cif
>>> cif.as_number('123')
123.0
>>> cif.as_int('123')
123
>>> cif.quote('two words')
"'two words'"
>>> cif.as_string(_)
'two words'
>>> cif.as_number(_)
nan
Reading and writing¶
Reading¶
We have a few reading functions that read a file (or a string, or a stream)
and return a document (DOM) – an instance of class Document.
#include <gemmi/read_cif.hpp>
// functions in namespace gemmi, from -lgemmi_cpp, usually linked with zlib or zlib-ng
// similar to cif::read_file, but uncompresses *.gz files on the fly
cif::Document read_cif_gz(const std::string& path, int check_level=1);
// reads the content of a CIF file from a memory buffer (name is used when reporting errors)
cif::Document read_cif_from_memory(const char* data, size_t size, const char* name, int check_level=1);
// reads from a string; it's in namespace gemmi::cif (for backward compatibility)
Document read_string(const std::string& data, int check_level=1);
#include <gemmi/cif.hpp>
// Header-only functions in namespace gemmi::cif.
// No linking, but slower compilation.
Document read_file(const std::string& filename, int check_level=1);
// name is used only when reporting errors.
Document read_memory(const char* data, const size_t size, const char* name, int check_level=1);
// Parameter bufsize determines the buffer size and only affects performance.
// These functions are slower than the ones above.
Document read_cstream(std::FILE *f, size_t bufsize, const char* name, int check_level=1);
Document read_istream(std::istream &is, size_t bufsize, const char* name, int check_level=1);
from gemmi import cif
# read and parse a CIF file; if the filename ends with .gz it is uncompressed on the fly
doc = cif.read_file('components.cif')
doc = cif.read_file('components.cif', check_level=1) # the same, 1 is the default
# the same as read_cif() except that it can also read mmJSON
doc = cif.read('components.cif')
# read content of a CIF file from string or bytes
doc = cif.read_string('data_this _is valid _cif content')
The optional check_level argument determines how strictly the CIF format
is checked (validated); see the list above.
The same checks can be run as separate low-level functions.
The read_file() call is equivalent to the following sequence:
doc = cif.Document()
doc.source = path
doc.parse_file(path)
if check_level > 0:
doc.check_for_missing_values()
doc.check_for_duplicates()
if check_level > 1:
for block in doc:
assert block.name != ' '
block.check_empty_loops(doc.source)
Similarly, the function read_string() can be replaced by a similar
sequence with Document.parse_string() doing the main job:
>>> doc = cif.Document()
>>> doc.parse_string('data_this _tag_has_no_value\n')
>>> doc[0].find_value('_tag_has_no_value')
''
>>> try: doc.check_for_missing_values()
... except RuntimeError: print('missing value')
...
missing value
As in other parts of Gemmi, functions that can read gzipped files can also
read from standard input. path specified as - means standard input.
If you’d have a file named -, use, for instance, path="./-".
Writing¶
A Document or Block can be written to a file or to a string.
Reading from a file and then writing to a file does not preserve whitespace.
The formatting of the output file is controlled by cif::WriteOptions,
which contains the following fields (by default, all are false or 0):
prefer_pairs(bool) – if set to true, write single-row loops as pairs,compact(bool) – if set to true, do not add blank lines between categories,misuse_hash(bool) – if set to true, put#(empty comments) between categories – the peculiar formatting used in the wwPDB archive; enables diff-ing (diff --ignore-space-change) with other such files,align_pairs(int) – pad tags in tag-value pairs to this width (if set to 33, values will be aligned at column 35, except where tags have more than 33 characters),align_loops(int) – if non-zero, columns in loops are aligned to the maximum string width in each column, but not more than this value; if one string in the column is very wide, that row will be misaligned, which is usually preferable to excessive padding.
#include <gemmi/to_cif.hpp>
// functions declared in namespace gemmi::cif
void write_cif_to_stream(std::ostream& os, const Document& doc, WriteOptions options);
void write_cif_block_to_stream(std::ostream& os, const Block& block, WriteOptions options);
>>> doc.write_file('1pfe-modified.cif')
>>> options = cif.WriteOptions()
>>> options.align_pairs = 33
>>> options.align_loops = 30
>>> doc.write_file('1pfe-aligned.cif', options)
>>> cif_in_string = doc.as_string(options)
>>> # Block also has methods write_file and as_string
>>> block_in_string = block.as_string(options)
JSON¶
cif::Document can be stored in a JSON format.
There are two semi-standards for mapping CIF to JSON:
mmJSON – specific to mmCIF and used by PDBj. In the mid-2010s each PDB site came up with a different format that’s more practical than mmCIF for use in WebGL molecular viewers:
RCSB announced MMTF, focused on minimizing file size and containing only essential information.
PDBe later came up with BinaryCIF, very similar to MMTF but somewhat bigger, keeping everything from mmCIF.
PDBj came up with mmJSON – not as compressed as the others, but doesn’t require custom parsers. Takes more disk space, but is much faster to parse. Sadly, it doesn’t have a specification and changes in the PDBj code that writes mmJSON can break things (it’s happened at least once).
CIF-JSON – agreed upon in an IUCr discussion group.
Neither mmJSON nor CIF-JSON is widely used.
Gemmi implementation of this feature predates CIF-JSON. We have a number of options that customize the translation. More details about it are given in the description of gemmi cif2json.
// see functions in:
#include <gemmi/json.hpp> // for reading
#include <gemmi/read_cif.hpp> // for reading possibly gzipped JSON files
#include <gemmi/to_json.hpp> // for writing
>>> doc = cif.read_string('data_this _is.minimal mmcif')
>>> doc.as_json(mmjson=True)
'{"data_this": {\n "is": {\n "minimal": ["mmcif"]\n }\n }\n}\n'
>>> cif.read_mmjson_string(_)
<gemmi.cif.Document with 1 blocks (this)>
>>> # functions cif.read_mmjson() and cif.read() read mmJSON files from disk
Binary serialization¶
This is the fastest way to serialize a Document or Block.
It is meant only for internal communication, between threads,
processes, or computers (client and server).
The format may change between releases.
It is based on the zpp serializer
library in C++ (see the header <gemmi/serializer>).
In Python, this format is used by Document’s and Block’s __getstate__
and __setstate__ functions,
which means it’s used for pickling and multiprocessing.
Document¶
Document contains Blocks, which correspond to CIF data blocks.
The blocks can be iterated over and accessed by index or by name
(each CIF block should have a unique name).
// You can directly access and modify the vector with Blocks
std::vector<Block> blocks;
// To access a block with known name use find_block().
// It returns nullptr if the block is not found.
Block* Document::find_block(const std::string& name)
>>> # Document can be iterated, accessed by block index and by block name:
>>> doc = cif.read_file("components.cif")
>>> len(doc)
25219
>>> doc[0]
<gemmi.cif.Block 000>
>>> doc[-1]
<gemmi.cif.Block ZZZ>
>>> doc['MSE']
<gemmi.cif.Block MSE>
>>> # The function doc.find_block(name) is like doc[name] ...
>>> doc.find_block('MSE')
<gemmi.cif.Block MSE>
>>> # ... except when the block is not found:
>>> doc.find_block('no such thing') # -> None
>>> # doc['no such thing'] # -> KeyError
As it is common for cif files to contain only a single block,
gemmi has a method sole_block() that returns the first block
if the document has only one block; otherwise it throws an exception.
// To access the only block in the file you may use:
Block& Document::sole_block()
>>> # Get the only block; throws exception if the document has more blocks.
>>> cif.read("../tests/1pfe.cif.gz").sole_block()
<gemmi.cif.Block 1PFE>
Document also has a member variable source that contains
the path of the file from which the document was read (if it was read
from a file).
>>> doc.source
'components.cif'
A new Document instance can be created with the default constructor.
To insert blocks, use:
// checks if the name is unique
Block& Document::add_new_block(const std::string& name, int pos=-1)
// or modify directly Document::blocks
# use one of two functions:
Document.add_new_block(name, pos=-1)
Document.add_copied_block(block, pos=-1)
As an example, here is how to start a new document:
>>> d = cif.Document()
>>> block_one = d.add_new_block('block-one')
>>> # populate block_one
To delete a block, in C++ access Document::blocks directly;
in Python use Document.__delitem__ (for example: del doc[1]).
Warning
Adding and removing blocks may invalidate references to other blocks in the same Document. This is expected when working with a C++ vector, but when using Gemmi from Python it is a flaw. The same applies to functions that add/remove items in a block. More precisely:
functions that add items (e.g.
add_new_block) may cause memory re-allocation invalidating references to all other items (blocks),functions that remove items (
__delitem__) invalidate references to all items after the removed one.
This means that you need to update a reference before using it:
block = doc[0] doc.add_new_block(...) # block gets invalidated block = doc[0] # block is valid again
Block¶
Each block has a name and a list of items. Each item is one of:
name-value pair (Pair)
table, a.k.a loop (Loop)
save frame (Block – the same data structure as for block)
A block headed by the word global_, part of the STAR syntax, although
not allowed
in CIF, is parsed into a Block with an empty name.
A block headed by bare data_, although allowed neither in CIF nor
in STAR, is parsed into a Block with name set to “ “ (the space character).
The name can be accessed as:
// member variable
std::string name;
>>> block = cif.read("../tests/1pfe.cif.gz").sole_block()
>>> block.name
'1PFE'
Items can be accessed as:
// member variable, a vector with Items.
// Item is implemented as an unrestricted union
// that holds one of Pair, Loop or Block.
std::vector<Item> items;
// Additionally, one may iterate over all Block's items,
// for example, here were are interested in "save frame":
for (cif::Item& item : block.items)
if (item.type == cif::ItemType::Frame)
// doing something with item.frame which is a (nested) Block
cif::Block& frame = item.frame;
>>> # Block can be iterated over, yielding items (class Item)
>>> for item in block:
... if item.line_number > 1670:
... if item.pair is not None:
... print('pair', item.pair)
... elif item.loop is not None:
... print('loop', item.loop)
... elif item.frame is not None:
... print('frame', item.frame)
...
pair ('_ndb_struct_conf_na.entry_id', '1PFE')
pair ('_ndb_struct_conf_na.feature', "'double helix'")
loop <gemmi.cif.Loop 8 x 25>
loop <gemmi.cif.Loop 5 x 43>
loop <gemmi.cif.Loop 3 x 3>
loop <gemmi.cif.Loop 83 x 10>
But this is not how we usually access items.
Usually, we use methods of Block that will be introduced further on.
Frame¶
(Very few people need it, skip this section.)
The named save frames (keyword save_) from the STAR specification
are used in CIF files only as sub-sections of a block.
The only place where they are encountered is mmCIF dictionaries.
The save-frame is stored as Block inside a Block and can be accessed with:
Block* Block::find_frame(std::string name)
>>> frame = block.find_frame('my_frame')
Pairs and Loops¶
The functions in this section can be considered low-level, because they are specific to either name-value pairs or to loops. The next sections introduce function that work with both pairs and loops.
Warning
Assuming that a certain value is contained in a pair, or that it is in a loop, is a common source of bugs when working with mmCIF files. To get the data, instead of using the functions in this section, it is recommended to use the abstractions (introduced in the next sections) that work regardless of whether the values are in pairs or loops.
For instance, when working with proteins one could assume that anisotropic ADP values are in a loop, but wwPDB also has entries with anisotropic ADP for only one atom – as name-value pairs. Conversely, one could think that R-free is always given as a name-value pair, but in entries from the joint X-ray and neutron refinement it is in a loop. Moreover, some CIF representations, such as mmJSON, do not preserve the distinction between pairs and loops at all.
A pair with a particular tag can be located using:
// Pair is simply defined as:
using Pair = std::array<std::string, 2>;
// returns nullptr if the tag is not found
const Pair* Block::find_pair(const std::string& tag) const
>>> block.find_pair('_cell.length_a')
('_cell.length_a', '39.374')
>>> block.find_pair('_no_such_tag') # return None
If you want just the value, you can use find_value().
This function also searches inside CIF loops and if there
is a matching tag with only a single value (which is semantically
equivalent to a name-value pair), that value is returned.
// returns nullptr if the tag is not found
const std::string* Block::find_value(const std::string& tag) const
>>> block.find_value('_cell.length_b')
'39.374'
>>> block.find_value('_cell.no_such_tag') # returns None
At last, if you’d like to access the Item containing the Pair
(for example, to check the line number), use the function:
// returns nullptr if the tag is not found
const Item* Block::find_pair_item(const std::string& tag) const
>>> item = block.find_pair_item('_cell.length_c')
>>> item.pair
('_cell.length_c', '79.734')
>>> item.line_number
72
>>> block.find_pair_item('_nothing') # return None
To add a name-value pair or modify an existing one, use:
void Block::set_pair(const std::string& tag, std::string value)
>>> block.set_pair('_year', '2030')
If the value needs quoting, the passed argument needs to be quoted,
for example, with cif::quote:
block.set_pair("_title", cif::quote("Goldilocks and the Three Bears"));
>>> block.set_pair('_title', cif.quote('Goldilocks and the Three Bears'))
If a new item is added, it is placed at the end of the block.
In mmCIF files, all name-value pairs in the same category must
be consecutive (an unwritten rule of the PDB).
You can move a newly added item to a different position with move_item(),
but that’s error-prone. Here is a trick to add a new pair
directly after the last item in a given category:
cif::ItemSpan(block, "_cell.").set_pair("_cell.length_a_esd", "?");
>>> # raw=True has the same meaning as in set_mmcif_category
>>> block.set_pairs('_cell.', {'length_a_esd': '?',
... 'length_b_esd': '?',
... 'length_c_esd': '?'}, raw=True)
Here is an example of how to create a CIF file from scratch:
>>> d = cif.Document()
>>> d.add_new_block('oak')
<gemmi.cif.Block oak>
>>> _.set_pair('_nut', 'acorn')
>>> print(d.as_string())
data_oak
_nut acorn
Loop is defined as two lists, tags and values:
struct Loop {
std::vector<std::string> tags;
std::vector<std::string> values;
// and a number of functions
};
In Python, loop.tags and loop.values return read-only lists.
They will be used in further examples.
To get values corresponding to a tag in a loop (table), you may use:
.. tab:: C++
Column Block::find_loop(const std::string& tag)
>>> block.find_loop('_atom_type.symbol')
<gemmi.cif.Column _atom_type.symbol length 6>
>>> list(_)
['C', 'CL', 'N', 'O', 'P', 'S']
Column, which is documented further on, has a method get_loop()
that gives access to the Loop.
Similarly to find_pair_item(), there is a function find_loop_item():
>>> block.find_loop_item('_atom_type.symbol')
<gemmi.cif.Item object at 0x...>
>>> _.loop
<gemmi.cif.Loop 6 x 1>
To add a row to an existing table (loop), use add_row():
>>> loop = block.find_loop('_atom_type.symbol').get_loop()
>>> loop.add_row(['Au'], pos=0)
>>> loop.add_row(['Zr']) # appends
>>> list(block.find_loop('_atom_type.symbol'))
['Au', 'C', 'CL', 'N', 'O', 'P', 'S', 'Zr']
add_row takes as an argument a list of strings, which should be quoted
if necessary. If you have a list of Python values use quote_list first:
>>> cif.quote_list([None, False, 3, -2.5, 'word', 'two words'])
['?', '.', '3', '-2.5', 'word', "'two words'"]
Columns can be added and removed:
>>> loop.add_columns(['_atom_type.description', '_atom_type.oxidation_number'], value='?')
>>> loop
<gemmi.cif.Loop 8 x 3>
>>> loop.remove_column('_atom_type.description')
>>> loop
<gemmi.cif.Loop 8 x 2>
set_all_values sets all the data in a table. It takes
a list of lists of strings. The lists of strings correspond to columns.
>>> loop = block.find_loop_item('_citation_author.citation_id').loop
>>> loop.tags
['_citation_author.citation_id', '_citation_author.name', '_citation_author.ordinal']
>>> loop.set_all_values([['primary']*2, [cif.quote('Alice A.'), cif.quote('Bob B.')], ['1', '2']])
>>> for row in block.find_mmcif_category('_citation_author.'):
... print(list(row))
['primary', "'Alice A.'", '1']
['primary', "'Bob B.'", '2']
In Python, individual values can be accessed with (row,column) tuples:
>>> loop[0,1]
"'Alice A.'"
>>> loop[0,1] = "'Carol C.'"
>>> loop.values # Loop.values is a read-only list (a copy of all values)
['primary', "'Carol C.'", '1', 'primary', "'Bob B.'", '2']
To add a new loop (replacing an old one if it exists) use init_loop
and populate it with tags and values:
Loop& Block::init_loop(const std::string& prefix, std::vector<std::string> tags)
// Then it can be populated by either setting directly tags and values,
// or by using Loop's methods such as add_row() or set_all_values().
>>> loop = block.init_loop('_ocean_', ['id', 'name'])
>>> # empty table is invalid in CIF, we need to add something
>>> loop.add_row(['1', cif.quote('Atlantic Ocean')])
In the above Python example, if the block already has tags _ocean_id
and/or _ocean_name and
if they are in a table: the table will be cleared and re-used,
if they are in name-value pairs: the pairs will be removed and a table will be created at the position of the first pair.
To reorder items, use block.move_item(old_pos, new_pos).
The current position of a tag can be obtained using block.get_index(tag).
>>> block.get_index('_entry.id')
0
>>> block.get_index('_ocean_id')
385
>>> block.move_item(0, -1) # move first item to the end
>>> block.get_index('_entry.id')
385
>>> block.get_index('_ocean_id')
384
>>> block.move_item(385, 0) # let's move it back (385 == -1 here)
To copy an item:
>>> item = block.find_loop_item('_ocean_id')
>>> new_block = cif.Block('new_block')
>>> new_block.add_item(item)
<gemmi.cif.Item object at 0x...>
Column¶
Column is a lightweight proxy class for working with both loop columns
and name-value pairs.
It was returned from find_loop above, but it is also returned from
a more general function find_values(), which searches for a given
tag in both loops and pairs.
C++¶
The C++ signature of find_values is:
Column Block::find_values(const std::string& tag)
Column has a few member functions:
// Number of rows in the loop. 0 means that the tag was not found.
int length() const;
// Returns pointer to the column name in the DOM.
// The name is the same as argument to find_loop() or find_values().
std::string* get_tag();
// Returns underlying Item (which contains either Pair or Loop).
Item* item();
// If it's in Loop, returns pointer to Loop; otherwise, nullptr.
Loop* get_loop() const;
// Get raw value (no bounds checking).
std::string& operator[](int n);
// Get raw value (after bounds checking).
std::string& at(int n);
// Short-cut for cif::as_string(column.at(n)).
std::string str(int n) const;
// Erases item for name-value pair; removes column for Loop
void erase();
Column also provides support for range-based for:
// mmCIF _chem_comp_atom is usually a table, but not always
for (const std::string &s : block.find_values("_chem_comp_atom.type_symbol"))
std::cout << s << std::endl;
If the column represents a name-value pair,
Column::get_loop() return nullptr
(and Column::get_length() returns 1).
Python¶
>>> block.find_values('_cell.length_a') # name-value pair
<gemmi.cif.Column _cell.length_a length 1>
>>> block.find_values('_atom_site.Cartn_x') # column in a loop
<gemmi.cif.Column _atom_site.Cartn_x length 342>
Column’s special method __bool__ tells if the tag was found.
__len__ returns the number of corresponding values.
__iter__, __getitem__ and __setitem__
get or set a raw string (i.e. string with quotes, if applicable).
As an example, let us shift all the atoms by 1A in the x direction:
>>> col_x = block.find_values('_atom_site.Cartn_x')
>>> for n, x in enumerate(col_x):
... col_x[n] = str(float(x) + 1)
To get the actual string content (without quotes) one may use
the method str:
>>> column = block.find_values('_chem_comp.formula')
>>> column[7]
"'H2 O'"
>>> column.str(7) # short-cut for cif.as_string(column[7])
'H2 O'
The tag is accessible (read/write) through the tag property:
>>> column.tag
'_chem_comp.formula'
If the tag was found in a loop, method get_loop returns a reference
to this Loop in the DOM. Otherwise it returns None.
>>> column.get_loop()
<gemmi.cif.Loop 12 x 7>
erase() removes the column from a loop, if this column is in a loop;
if it’s a tag-value pair it erases the containing item.
>>> loop = column.get_loop()
>>> column.erase()
>>> loop # one column less
<gemmi.cif.Loop 12 x 6>
Table¶
Usually we want to access multiple columns at once,
so the library has another abstraction (Table)
that can be used with multiple tags.
Table is returned by Block.find().
Like Column, it is a lightweight, iterable view of the data,
but it is for querying multiple related tags at the same time.
The first form of find() takes a list of tags:
Table Block::find(const std::vector<std::string>& tags)
// example:
block.find({"_entity_poly_seq.entity_id", "_entity_poly_seq.num", "_entity_poly_seq.mon_id"});
>>> block.find(['_entity_poly_seq.entity_id', '_entity_poly_seq.num', '_entity_poly_seq.mon_id'])
<gemmi.cif.Table 18 x 3>
Since tags in one loop tend to have a common prefix (category name), the library also provides a second form that takes the common prefix as the first argument:
Table Block::find(const std::string& prefix, const std::vector<std::string>& tags)
// example:
block.find("_entity_poly_seq.", {"entity_id", "num", "mon_id"});
>>> block.find('_entity_poly_seq.', ['entity_id', 'num', 'mon_id'])
<gemmi.cif.Table 18 x 3>
Note that find is not aware of dictionaries and categories,
so the category name should end with a separator
(dot for mmCIF files, as shown above).
In the example above, all the tags are required. If one of them is absent,
the returned Table is empty.
>>> block.find('_entity_poly_seq.', ['entity_id', 'num', 'non_existent'])
<gemmi.cif.Table nil>
Tags (all except the first one) can be marked
as optional by adding prefix ?:
Table table = block.find({"_required_tag", "?_optional_tag"})
>>> table = block.find(['_required_tag', '?_optional_tag'])
In such case the returned table may contain either one or two columns.
Before accessing the column corresponding to an optional tag one must check
if the column exists using Table::has_column() (or, alternatively,
the equivalent Table::Row::has() which will be introduced later):
bool Table::has_column(int n) const
>>> block.find('_entity_poly_seq.', ['entity_id', '?num', '?bleh'])
<gemmi.cif.Table 18 x 3>
>>> _.has_column(0), _.has_column(1), _.has_column(2)
(True, True, False)
The Table has functions to check its shape:
bool ok() const; // true if the table is not empty
size_t width() const; // number of columns
size_t length() const; // number of rows
>>> table = block.find('_entity_poly_seq.', ['entity_id', 'num', 'mon_id'])
>>> # instead of ok() in Python we use __bool__()
>>> assert table, "table.__bool__() is expected to return True"
>>> table.width() # number of columns
3
>>> len(table) # number of rows
18
If the Table abstracts a loop, the Loop class can be accessed using:
Loop* get_loop(); // nullptr for tag-value pairs
>>> table.loop # None for tag-value pairs
<gemmi.cif.Loop 18 x 4>
Tag-value pairs can be converted to a loop using:
>>> table.ensure_loop()
but make sure to call it only when the table represents the whole category (see example below).
If a prefix was specified when calling find, the prefix length is stored and the prefix can be retrieved:
size_t prefix_length;
std::string get_prefix() const;
>>> table.prefix_length
17
>>> table.get_prefix()
'_entity_poly_seq.'
Table also has the function erase() that deletes all tags and data
associated with the table.
See an example in the CCD section below.
Row-wise access¶
Most importantly, the Table provides access to data in rows and columns.
We can get a row (Table::Row) that in turn provides access to value strings:
Row Table::operator[](int n) // access Row
Row Table::at(int n) // the same but with bounds checking
// and also begin(), end() and iterator that enable range-for.
// Returns the first row that has the specified string in the first column.
Row Table::find_row(const std::string& s)
// Makes sure that the table has only one row and returns it.
Row Table::one()
>>> table[0]
<gemmi.cif.Table.Row: 1 1 DG>
>>> # and of course it's iterable
>>> for row in table: pass
>>> # Returns the first row that has the specified string in the first column.
>>> table.find_row('2')
<gemmi.cif.Table.Row: 2 1 DSN>
as well as to the tags:
Row tags(); // pseudo-row that contains tags
>>> table.tags
<gemmi.cif.Table.Row: _entity_poly_seq.entity_id _entity_poly_seq.num _entity_poly_seq.mon_id>
Tags in the table (in the C++ layer) are references to tags in the CIF block. If the requested tags are not found, the returned nil table has no tags. Such table has 0 rows, but it can be iterated like an empty list:
>>> block.find(['_absent_tag']).tags
<gemmi.cif.Table.Row:>
>>> for row in block.find(['_absent_tag']): print(row)
... # nothing gets printed, but there is no error
Table::Row has functions for accessing the values:
// Get raw value.
std::string& Table::Row::operator[](int n) // no bounds checking
std::string& Table::Row::at(int n) // with bounds checking
// and also begin(), end(), iterator, const_supports iterators.
>>> for row in table: print(row[-1], end=',')
DG,DC,DG,DT,DA,DC,DG,DC,DSN,ALA,N2C,NCY,MVA,DSN,ALA,NCY,N2C,MVA,
>>>
>>> row = table[9]
>>> for value in row: print(value, end=',')
2,2,ALA,
>>> row[2]
'ALA'
>>> row[-1] # the same, because this table has two rows
'ALA'
>>> row.get(2) # the same, but would return None instead of IndexError
'ALA'
>>> row['mon_id'] # the same, but slightly slower than numeric index
'ALA'
>>> row['_entity_poly_seq.mon_id'] # the same
'ALA'
and a few convenience functions, including:
size_t Table::Row::size() const // the width of the table
std::string Table::Row::str(int n) const // short-cut for cif::as_string(row.at(n))
bool Table::Row::has(int n) const // the same as Table::has_column(n)
>>> len(row) # the same as table.width()
3
>>> row.str(2) # if the value is in quotes, it gets un-quoted
'ALA'
>>> row.has(2) # the same as table.has_column(2)
True
and a property:
int row_index
>>> row.row_index
9
Individual values in a row can be directly modified. This includes tags, which are accessible as a special row. As an example, let us swap two names (these two tend to have identical values, so no one will notice):
cif::Table table = block.find("_atom_site.", {"label_atom_id", "auth_atom_id"});
cif::Table::Row tags = table.tags();
std::swap(tags[0], tags[1]);
>>> tags = block.find('_atom_site.', ['label_atom_id', 'auth_atom_id']).tags
>>> tags[0], tags[1] = tags[1], tags[0]
Function Table::append_row appends a row.
It won’t work if the table is composed of tag-value pairs (which isn’t
the case here, but let’s make this example as general as possible),
so we call Table::ensure_loop() first.
This function, in turn, won’t work well if the table contains only a subset
of the category, thus find_mmcif_category():
>>> block.find_mmcif_category('_entity_poly_seq.').ensure_loop()
>>> table.append_row(['3', '4', 'new'])
>>> table[-1]
<gemmi.cif.Table.Row: 3 4 new>
>>> _.row_index
18
If new tags are added to this category in the future,
the corresponding values in appended rows will be filled with ..
We can also move a row to a different position:
>>> table.move_row(old_pos=-1, new_pos=0) # move the last row to the front
>>> table[0]
<gemmi.cif.Table.Row: 3 4 new>
and remove a row from Table (function Table::remove_row):
>>> table.remove_row(18) # or: del table[18]
To efficiently remove multiple consecutive rows use
C++ function Table::remove_rows or Python __delitem__ with slice:
>>> del table[12:15]
As is usual with any containers (in both Python and C++), if you want to remove items while iterating over them, the iteration must be done backward. Here is an example that removes atoms with zero occupancy:
>>> doc = cif.read('../tests/3dg1_final.cif')
>>> atom_table = doc[0].find(['_atom_site.occupancy'])
>>> for i in range(len(atom_table)-1, -1, -1):
... if float(atom_table[i][0]) == 0:
... del atom_table[i]
...
>>> doc.write_file('out.cif')
Column-wise access¶
Table gives also access to columns, represented by the previously
introduced Column:
Column Table::column(int index)
// alternatively, specify tag name
Column Table::find_column(const std::string& tag)
>>> table.column(0)
<gemmi.cif.Column _entity_poly_seq.entity_id length 15>
>>> table.find_column('_entity_poly_seq.mon_id')
<gemmi.cif.Column _entity_poly_seq.mon_id length 15>
If the table is created in a function that uses prefix,
the prefix can be omitted in find_column:
Table t = block.find("_entity_poly_seq.", {"entity_id", "num", "mon_id"});
Column col = t.find_column(2);
// is equivalent to
Column col = t.find_column("_entity_poly_seq.mon_id");
// is equivalent to
Column col = t.find_column("mon_id");
>>> table.find_column('mon_id')
<gemmi.cif.Column _entity_poly_seq.mon_id length 15>
C++ note:
both Column and Table::Row have functions begin() and end()
in const and non-const variants, returning iterator and
const_iterator types, respectively. These types satisfy requirements
of the BidirectionalIterator concept.
Conversely, the iterator over the rows of Table is a minimalistic
structure – just enough get the range-for work.
mmCIF categories¶
mmCIF files group data into categories. All mmCIF tags have a dot
(e.g. _entry.id) and the category name is the part before the dot.
We have two functions to work with categories. One returns a list of all categories in the block:
std::vector<std::string> Block::get_mmcif_category_names() const
>>> block.get_mmcif_category_names()[:3]
['_entry.', '_audit_conform.', '_database_2.']
The other returns a Table with all tags (and values) belonging to
the specified category:
Table Block::find_mmcif_category(std::string cat)
>>> block.find_mmcif_category('_entry.')
<gemmi.cif.Table 1 x 1>
>>> _.tags[0], _[0][0]
('_entry.id', '1PFE')
>>> cat = block.find_mmcif_category('_database_2.')
>>> cat
<gemmi.cif.Table 4 x 2>
>>> list(cat.tags)
['_database_2.database_id', '_database_2.database_code']
>>> for row in cat:
... print('%s: %s' % tuple(row))
PDB: 1PFE
NDB: DD0057
RCSB: RCSB019291
WWPDB: D_1000019291
>>> cat[3][1]
'D_1000019291'
Additionally, Python bindings have functions get_mmcif_category
and set_mmcif_category that translate between an mmCIF category
and Python dictionary:
>>> block.get_mmcif_category('_entry.')
{'id': ['1PFE']}
>>> sorted(block.get_mmcif_category('_database_2.').keys())
['database_code', 'database_id']
? and . are translated to None and False.
To disable this translation and get “raw” strings, add argument raw=True.
>>> default = block.get_mmcif_category('_software')
>>> raw = block.get_mmcif_category('_software', raw=True)
>>> for name in ['name', 'version', 'citation_id']:
... print(default[name][0], raw[name][0])
...
EPMR EPMR
False .
None ?
set_mmcif_category() takes a category name and a dictionary (that
maps tags to lists) and creates or replaces the corresponding category:
>>> ocean_data = {'id': [1, 2, 3], 'name': ['Atlantic', 'Pacific', 'Indian']}
>>> block.set_mmcif_category('_ocean', ocean_data)
It also takes the optional raw parameter (default: False).
Specify raw=True if the values are already quoted, otherwise
they will be quoted twice.
>>> block.set_mmcif_category('_raw', {'a': ['?', '.', "'a b'"]}, raw=True)
>>> list(block.find_values('_raw.a'))
['?', '.', "'a b'"]
>>> block.set_mmcif_category('_nop', {'a': ['?', '.', "'a b'"]}, raw=False)
>>> list(block.find_values('_nop.a'))
["'?'", "'.'", '"\'a b\'"']
>>> block.set_mmcif_category('_ok', {'a': [None, False, 'a b']}, raw=False)
>>> list(block.find_values('_ok.a'))
['?', '.', "'a b'"]
Finally, we have a variant of init_loop for working with mmCIF categories:
>>> loop = block.init_mmcif_loop('_ocean.', ['id', 'name'])
>>> loop.add_row(['1', 'Atlantic'])
The subtle difference from generic init_loop is that if the block
has other name-value pairs in the same category (say, _ocean.depth 8.5)
init_loop leaves them untouched, but init_mmcif_loop removes them.
Additionally, like in other _mmcif_ functions, the trailing dot
in the category name may be omitted (but the leading underscore is required).
Validation¶
A CIF document can conform to a dictionary (ontology, think DTD for XML or JSON Schema for JSON). A dictionary, written in one of the versions of DDL (Dictionary Definition Language), is itself a CIF document. There are three versions of DDL:
DDL1 is the simplest. It is used, for instance, for small molecule CIFs.
DDL2 is used for PDBx/mmCIF, with activity in this area centered around the PDB.
DDLm is a newer version (from around 2011) from the IUCr’s COMCIFS (Committee for the Maintenance of the CIF Standard). It’s not widely used yet and, like CIF2, is not supported by Gemmi.
Gemmi is primarily used in structural biology and is mostly exercised with mmCIF and DDL2. DDL1 is supported to a limited extent (which could be expanded if there was a good use case).
Note
In most cases, it’s simpler to use the command-line program gemmi validate instead of the functions described below. If you use mmCIF-like files, make sure you read notes about DDL2.
The validation capabilities are implemented in class cif::Ddl.
Let’s start with a simple example, a pet weighting experiment:
>>> pet_example = '''\
... data_pets
... loop_
... _pet_id
... _pet_species
... _pet_weight
... 1 parrot 2
... 2 dog 15
... '''
Now let’s create a contrived DDL1 dictionary for it:
>>> pet_ddl = cif.read_string('''\
... data_pet_index
... _name '_pet_id'
... _category pet
... _type numb
...
... data_pet_species
... _name '_pet_species'
... _category pet
... _type char
... loop_ _enumeration parrot cat dog
...
... data_pet_weight
... _name '_pet_weight'
... _category pet
... _type numb
... _enumeration_range 0.0:100.0
... _units kg
... ''')
The Ddl class must be first supplied with a dictionary and can then validate
CIF files.
>>> validator = cif.Ddl(logger=sys.stdout)
>>> validator.read_ddl(pet_ddl)
>>> validator.validate_cif(cif.read_string(pet_example))
True
Now let’s append a line that will trigger errors:
>>> pet_example += '''
... 3 hippo 3000
... '''
>>> validator.validate_cif(cif.read_string(pet_example))
string:2 [pets] _pet_species: hippo is not one of the allowed values:
parrot
cat
dog
string:2 [pets] _pet_weight: value out of expected range: 3000
False
Errors are sent to a logger as described in a separate section. The logger is set in the constructor and can be changed at any point:
>>> validator.set_logger((None, 0))
>>> validator.validate_cif(cif.read_string(pet_example))
False
Calling read_ddl() moves the content of a Document to the Ddl class,
leaving the original object empty (it’s slightly faster this way).
read_ddl() can be called multiple times to use multiple dictionaries
(or extensions) simultaneously.
Ddl has a few flags to enable or disable certain types of checks.
These correspond to the optional checks listed in the documentation
of the gemmi validate subcommand.
In C++, these are member variables that can be set directly.
In Python, they are set through keyword arguments in the constructor
(except for use_deposition_checks, which is set directly).
The minimal example above used a contrived dictionary. Normally, you will
use a dictionary downloaded from the IUCr, wwPDB or another source –
perhaps with your own extensions. So you’ll use cif.read() instead of
cif.read_string().
Notes on DDL2¶
The commonly used DDL2-based dictionaries are
available from wwPDB.
To validate mmCIF files, use the current version of the PDBx/mmCIF
dictionary (mmcif_pdbx_v50.dic as of 2025).
The original IUCr mmCIF dictionary (cif_mm.dic) is now only of historical
interest. It was actively developed in the 1990s, but in the 2000s development
was taken over by the PDB under the PDBx/mmCIF name. Formally, PDBx/mmCIF
is an extension of the IUCr mmCIF, but for all practical purposes it can be
thought of as the current mmCIF dictionary.
When we talk about mmCIF files, it’s shorthand for PDBx/mmCIF
or mmCIF-like files. No software targets the original mmCIF specification.
The mmCIF dictionary itself is massive—over 5MB of text—so it, too, can
use some validation. That’s what the mmcif_ddl.dic (DDL2) dictionary is for.
This dictionary can also validate itself, closing the loop:
$ gemmi validate -d mmcif_ddl.dic mmcif_ddl.dic
The PDBx/mmCIF dictionary can be used to validate coordinate,
structure factor and chemical component files.
From a validation perspective, they are all the same thing.
That’s why all categories in mmCIF files are, according to the dictionary,
optional (_category.mandatory_code no) – we can’t tell what the file
must contain. However, many items are marked as mandatory within
categories (_item.mandatory_code). If you’ve ever wondered
about the difference between null values ? and . in mmCIF files:
the PDB’s software writes ? and . for optional and mandatory items,
respectively (an implementation detail that deviates from the CIF 1.1 spec).
If an item is mandatory, it only means that if its category is present,
the tag must also be present, but its value can be unknown or n/a.
DDL2 is missing a comprehensive specification. What is not covered in
International Tables for Crystallography, vol G (2006), has to be
inferred from studying dictionaries and asking around.
Parent-child relationships are particularly challenging.
Tags may have associated parent tags (e.g. _entity.id is the parent
of _entity_poly.entity_id), and groups of tags may have associated
parent groups (defined in the pdbx_item_linked_group category).
But it’s unclear if every parent must exist.
The PDB’s own validation software (CifCheck from
mmcif-dict-suite)
checks for the presence of parent tags but has a long list of arbitrary
exceptions hardcoded into the program, otherwise most of the files from
the PDB wouldn’t validate. In gemmi, the relationships are not checked
by default, but there is an option for it (-p in gemmi validate).
In some cases, broken relationships are fixable. In others, there is a fundamental mismatch between the design of the mmCIF schema and the capabilities of DDL2. For example, some aspects of polymers and non-polymers are described in different categories, but a residue can’t be conditionally linked to one or the other. So, it’s linked only to polymeric categories, leaving the schema partially incorrect.
In addition to _item_type (data type), the PDBx/mmCIF spec also features
_pdbx_item_type (described as an “alternate data type”).
Similarly, _item_range (permissible range) has its twin
_pdbx_item_range (alternate permissible range), and
_item_enumeration has the alternate _pdbx_item_enumeration.
While the purpose of this dualism is not documented, the alternates are
interpreted as deposition constraints (typically, stricter checks).
Gemmi provides the use_deposition_checks option (--depo in the command-line
program) to apply the alternate criteria instead of the standard ones.
If you run gemmi validation in verbose mode, you might see warnings about incorrect regular expressions in a dictionary. In general, regexes come in various flavors. Over the years, some flavors have been formally defined and standardized (POSIX BRE, ERE, RegExp in EcmaScript, etc.). I think the regexes used in DDL2 are closest to POSIX ERE (Extended RegExp). Gemmi has hacks for parsing all the regexes that have been in mmCIF dictionaries for a long time, but sometimes new ones are added that are inconsistent with the older ones, so full support can’t be guaranteed.
Dictionaries allow only relatively simple checks. When you deposit files to the PDB, they are primarily validated by other means. Coordinate files are processed by a program called MAXIT, and structure factor files – by SF-CONVERT. These are part of a C++ codebase that has been developed at RCSB since the late 1990s. The files you deposit don’t need to strictly conform to the dictionary, but they must be able to pass through the processing programs, which rewrite them anyway. Attempts to make an mmCIF file more conformant with the spec sometimes backfire, choking the OneDep pipeline.
Validation helps spot certain types of mistakes but shouldn’t be overemphasized. When generating an mmCIF file, the goal is to ensure that it can be read and correctly interpreted by the software it will be used with next. Validation against a dictionary is a guideline, not the goal.
Design choices¶
Parser¶
Parsing formal languages is a well-researched topic in computer science. The first versions of lex and yacc - popular tools that generate lexical analyzers and parsers - were written in 1970’s. Today many tools exist to translate grammar rules into C/C++ code. On the other hand many compilers and high-profile tools use hand-coded parsers - as it is more flexible.
Looking at other STAR and CIF parsers – some use parser generators (COD::CIF::Parser and starlib2 use yacc/bison, iotbx.cif uses Antlr3), others are hand-coded.
I had experience with flex/bison and Boost.Spirit (and I wanted to try also Lemon and re2c) but I decided to use PEGTL for this task (and I’m very happy with this choice). I was convinced by the TAOC++ JSON parser that is based on PEGTL and has a good balance of simplicity and performance.
PEGTL is a C++ library (not a generator) for creating PEG parsers. PEG stands for Parsing Expression Grammar – a simpler approach than the traditional Context Free Grammar.
As a result, our parser depends on a third-party (header-only) library, but the parser itself is pretty simple.
And it is still the fastest open-source CIF parser (at least in the hands of the author). While further improvement would be possible (some JSON parsers are much faster) it is not a priority, the parser is fast enough.
Data structures¶
The next thing is how we store the data read from file. We decided to rely on the C++ standard library where we can.
Generally, storage of such data involves (in C++ terms) some containers and a union/variant type for storing values of mixed types.
We use primarily std::vector as a container.
A custom structure Item with (unrestricted) union is used as
a variant-like class.
Strings are stored in std::string and it is fast enough.
Mainstream C++ standard libraries have short string optimization (SSO)
for up to 15 or 22 characters, which covers most of the values in mmCIF files.
Examples¶
The examples here use C++ or Python. Full working code can be found in the examples directory.
mmCIF to XYZ¶
To start with something simple, let us convert mmCIF to the XYZ format:
#include <iostream>
#include <gemmi/cif.hpp>
#include <gemmi/util.hpp> // for gemmi::join_str
//#include <boost/algorithm/string/join.hpp> // for boost::algorithm::join
void convert_to_xyz(cif::Document doc) {
cif::Table atoms = doc.sole_block().find("atom_site_.",
{"type_symbol", "Cartn_x", "Cartn_y", "Cartn_z"});
std::cout << atoms.length() << "\n\n";
for (auto row : atoms)
std::cout << gemmi::join_str(row, " ") << "\n";
//std::cout << boost::algorithm::join(row, " ") << "\n";
}
mmJSON-like data¶
Gemmi has a built-in support for mmJSON and comes with converters cif2json and json2cif, but just as an exercise let us convert mmJSON to mmCIF in Python:
import json
import sys
from gemmi import cif
file_in, file_out = sys.argv[1:]
with open(file_in) as f:
json_data = json.load(f)
assert len(json_data) == 1 # data_1ABC
(block_name, block_data), = json_data.items()
assert block_name.startswith('data_')
# Now block_data is a dictionary that maps category names to dictionaries
# that in turn map column names to lists with values.
doc = cif.Document()
block = doc.add_new_block(block_name[5:])
for cat, data in block_data.items():
block.set_mmcif_category('_'+cat, data)
doc.write_file(file_out)
Analysing the PDB archive¶
The examples here can be run on one or more PDBx/mmCIF files. The ones that perform PDB-wide analysis are meant to be run on a local copy of the mmCIF archive (gzipped, don’t uncompress!).
Some of the exercises analysing the PDB archive were overdone, ending up as visualization projects.
auth vs label¶
When you look at the list of atoms (_atom_site.*) in mmCIF files
some columns seem to be completely redundant. Are they?
| group_PDB | id | type_symbol | label_atom_id | label_alt_id | label_comp_id | label_asym_id | label_entity_id | label_seq_id | pdbx_PDB_ins_code | Cartn_x | Cartn_y | Cartn_z | occupancy | B_iso_or_equiv | Cartn_x_esd | Cartn_y_esd | Cartn_z_esd | occupancy_esd | B_iso_or_equiv_esd | pdbx_formal_charge | auth_seq_id | auth_comp_id | auth_asym_id | auth_atom_id | pdbx_PDB_model_num |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATOM | 1 | N | N | . | GLY | A | 1 | 1 | ? | -9.009 | 4.612 | 6.102 | 1.00 | 16.77 | ? | ? | ? | ? | ? | ? | 1 | GLY | A | N | 1 |
| ATOM | 2 | C | CA | . | GLY | A | 1 | 1 | ? | -9.052 | 4.207 | 4.651 | 1.00 | 16.57 | ? | ? | ? | ? | ? | ? | 1 | GLY | A | CA | 1 |
| ATOM | 3 | C | C | . | GLY | A | 1 | 1 | ? | -8.015 | 3.140 | 4.419 | 1.00 | 16.16 | ? | ? | ? | ? | ? | ? | 1 | GLY | A | C | 1 |
| ATOM | 4 | O | O | . | GLY | A | 1 | 1 | ? | -7.523 | 2.521 | 5.381 | 1.00 | 16.78 | ? | ? | ? | ? | ? | ? | 1 | GLY | A | O | 1 |
| ATOM | 5 | N | N | . | ASN | A | 1 | 2 | ? | -7.656 | 2.923 | 3.155 | 1.00 | 15.02 | ? | ? | ? | ? | ? | ? | 2 | ASN | A | N | 1 |
| ATOM | 6 | C | CA | . | ASN | A | 1 | 2 | ? | -6.522 | 2.038 | 2.831 | 1.00 | 14.10 | ? | ? | ? | ? | ? | ? | 2 | ASN | A | CA | 1 |
| ATOM | 7 | C | C | . | ASN | A | 1 | 2 | ? | -5.241 | 2.537 | 3.427 | 1.00 | 13.13 | ? | ? | ? | ? | ? | ? | 2 | ASN | A | C | 1 |
| ATOM | 8 | O | O | . | ASN | A | 1 | 2 | ? | -4.978 | 3.742 | 3.426 | 1.00 | 11.91 | ? | ? | ? | ? | ? | ? | 2 | ASN | A | O | 1 |
| ATOM | 9 | C | CB | . | ASN | A | 1 | 2 | ? | -6.346 | 1.881 | 1.341 | 1.00 | 15.38 | ? | ? | ? | ? | ? | ? | 2 | ASN | A | CB | 1 |
| ATOM | 10 | C | CG | . | ASN | A | 1 | 2 | ? | -7.584 | 1.342 | 0.692 | 1.00 | 14.08 | ? | ? | ? | ? | ? | ? | 2 | ASN | A | CG | 1 |
| ATOM | 11 | O | OD1 | . | ASN | A | 1 | 2 | ? | -8.025 | 0.227 | 1.016 | 1.00 | 17.46 | ? | ? | ? | ? | ? | ? | 2 | ASN | A | OD1 | 1 |
| ATOM | 12 | N | ND2 | . | ASN | A | 1 | 2 | ? | -8.204 | 2.155 | -0.169 | 1.00 | 11.72 | ? | ? | ? | ? | ? | ? | 2 | ASN | A | ND2 | 1 |
| ... | |||||||||||||||||||||||||
| ATOM | 58 | O | OH | . | TYR | A | 1 | 7 | ? | 3.766 | 0.589 | 10.291 | 1.00 | 14.39 | ? | ? | ? | ? | ? | ? | 7 | TYR | A | OH | 1 |
| ATOM | 59 | O | OXT | . | TYR | A | 1 | 7 | ? | 11.358 | 2.999 | 7.612 | 1.00 | 17.49 | ? | ? | ? | ? | ? | ? | 7 | TYR | A | OXT | 1 |
| HETATM | 60 | O | O | . | HOH | B | 2 | . | ? | -6.471 | 5.227 | 7.124 | 1.00 | 22.62 | ? | ? | ? | ? | ? | ? | 8 | HOH | A | O | 1 |
| HETATM | 61 | O | O | . | HOH | B | 2 | . | ? | 10.431 | 1.858 | 3.216 | 1.00 | 19.71 | ? | ? | ? | ? | ? | ? | 9 | HOH | A | O | 1 |
| HETATM | 62 | O | O | . | HOH | B | 2 | . | ? | -11.286 | 1.756 | -1.468 | 1.00 | 17.08 | ? | ? | ? | ? | ? | ? | 10 | HOH | A | O | 1 |
It is hard to manually find an example
where _atom_site.auth_atom_id differs from _atom_site.label_atom_id, or
where _atom_site.auth_comp_id differs from _atom_site.label_comp_id.
So here is a small C++ program:
// Compare pairs of columns from the _atom_site table.
#include <gemmi/read_cif.hpp>
#include <gemmi/cif.hpp>
#include <iostream>
#include <filesystem> // requires C++17
namespace fs = std::filesystem;
namespace cif = gemmi::cif;
void print_differences(cif::Block& block, const std::string& name) {
for (auto row : block.find("_atom_site.", {"auth_" + name, "label_" + name}))
if (row[0] != row[1])
std::cout << block.name << ": " << name << " "
<< row[0] << " -> " << row[1] << std::endl;
}
bool ends_with(const std::string& str, const std::string& suffix) {
size_t sl = suffix.length();
return str.length() >= sl && str.compare(str.length() - sl, sl, suffix) == 0;
}
int main(int argc, char* argv[]) {
if (argc != 2)
return 1;
for (auto& p : fs::recursive_directory_iterator(argv[1])) {
std::string path = p.path().u8string();
cif::Document doc = gemmi::read_cif_gz(path);
// What author's atom names were changed?
print_differences(doc.sole_block(), "atom_id");
// What author's residue names were changed?
print_differences(doc.sole_block(), "comp_id");
}
return 0;
}
We compile it, run it, and come back after an hour:
$ g++ -O2 -Iinclude examples/auth_label.cpp src/gz.cpp -lz
$ ./a.out pdb_copy/mmCIF
3D3W: atom_id O1 -> OD
1TNI: atom_id HN2 -> HN3
1S01: comp_id CA -> UNL
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
2KI7: atom_id H -> H1
4E1U: atom_id H101 -> H103
4WH8: atom_id H3 -> H391
4WH8: atom_id H4 -> H401
3V2I: atom_id UNK -> UNL
1AGG: atom_id H3 -> H
1AGG: atom_id H3 -> H
1AGG: atom_id H3 -> H
Update: the result above is from 2017. In the meantime, the PDB removed the differences. Tags auth_atom_id and auth_comp_id are now completely redundant (they always have been, except for mistakes).
Amino acid frequency¶
After seeing a paper in which amino acid frequency was averaged over 5000+ proteomes (Ala 8.76%, Cys 1.38%, Asp 5.49%, etc) we may want to compare it with the frequency in the PDB database. So we write a little script
#!/usr/bin/env python
# Check amino-acid frequency in the PDB database (or it's subset)
# by reading meta-data from mmCIF files.
import sys
from collections import Counter
from gemmi import cif, CifWalk
totals = Counter()
for arg in sys.argv[1:]:
for path in CifWalk(arg, try_pdbid='M'):
# read file (uncompressing on the fly) and get the only block
block = cif.read(path).sole_block()
# find table with the sequence
seq = block.find('_entity_poly_seq.', ['entity_id', 'mon_id'])
# convert table with chain types (protein/DNA/RNA) to dict
entity_types = dict(block.find('_entity_poly.', ['entity_id', 'type']))
# and count these monomers that correspond to a protein chain
aa_counter = Counter(row.str(1) for row in seq
if 'polypeptide' in entity_types[row.str(0)])
totals += aa_counter
# print residue counts for each file
print(block.name, *(f'{m}:{c}' for (m, c) in aa_counter.most_common()))
# finally, print the total counts as percentages
f = 100.0 / sum(totals.values())
print('TOTAL', *(f'{m}:{c*f:.2f}%' for (m, c) in totals.most_common(20)))
We can run this script on our copy of the PDB database (mmCIF format) and get such an output:
200L ALA:17 LEU:15 LYS:13 ARG:13 THR:12 ASN:12 GLY:11 ILE:10 ASP:10 VAL:9 GLU:8 SER:6 TYR:6 GLN:5 PHE:5 MET:5 PRO:3 TRP:3 HIS:1
...
4ZZZ LEU:40 LYS:33 SER:30 ASP:25 GLY:25 ILE:25 VAL:23 ALA:19 PRO:18 GLU:18 ASN:17 THR:16 TYR:16 GLN:14 ARG:11 PHE:10 HIS:9 MET:9 CYS:2 TRP:2
TOTAL LEU:8.90% ALA:7.80% GLY:7.34% VAL:6.95% GLU:6.55% SER:6.29% LYS:6.18% ASP:5.55% THR:5.54% ILE:5.49% ARG:5.35% PRO:4.66% ASN:4.16% PHE:3.88% GLN:3.77% TYR:3.45% HIS:2.63% MET:2.14% CYS:1.38% TRP:1.35%
On my laptop it takes about an hour, using a single core. Most of this hour is spent on tokenizing the CIF files and copying the content into a DOM structure, what could be largely avoided given that we use only sequences not atoms. But it is not worth the effort to optimize one-off scripts. The same goes for using multiple processor cores.
Custom PDB search¶
We may need to go through a local copy of the PDB archive to find entries according to criteria that cannot be queried in RCSB/PDBe/PDBj web interfaces. For example, if for some reason we need PDB entries with large number of anisotropic B-factors, we may write a quick script:
#!/usr/bin/env python
"Find PDB entries with more than 50,000 anisotropic B-factors."
import sys
from gemmi import cif, CifWalk
for path in CifWalk(sys.argv[1]):
block = cif.read(path).sole_block()
anis = block.find_values("_atom_site_anisotrop.id")
if len(anis) > 50000:
print(block.name, len(anis))
and then run it for an hour or so.
$ ./examples/simple_search.py pdb_copy/mmCIF
5AEW 60155
5AFI 98325
3AIB 52592
...
Solvent content vs resolution¶
Let’s say we want to generate a plot of solvent content as a function of dmin, similar to the plots by C. X. Weichenberger and B. Rupp in Acta Cryst D and CNN), but less smoothed and with duplicate entries included.
In macromolecular crystallography solvent content is conventionally estimated as:
VS = 1 – 1.230 / VM
where VM is the Matthews coefficient defined as VM=V/m (volume of the asymmetric unit over the molecular weight of all molecules in this volume).
Weichenberger and Rupp calculated VS themselves, but we will simply use the values present in mmCIF files. So first we extract VM, VS and dmin, as well as number of DNA/RNA chains (protein-only entries will have 0 here), deposition date and group ID (which will be explained later).
def gather_data():
'read mmCIF files and write down a few numbers (one file -> one line)'
writer = csv.writer(sys.stdout, dialect='excel-tab')
writer.writerow(['code', 'na_chains', 'vs', 'vm', 'd_min', 'date', 'group'])
for path in get_file_paths_from_args():
block = cif.read(path).sole_block()
code = cif.as_string(block.find_value('_entry.id'))
na = sum('nucleotide' in t[0]
for t in block.find_values('_entity_poly.type'))
vs = block.find_value('_exptl_crystal.density_percent_sol')
vm = block.find_value('_exptl_crystal.density_Matthews')
d_min = block.find_value('_refine.ls_d_res_high')
dep_date_tag = '_pdbx_database_status.recvd_initial_deposition_date'
dep_date = parse_date(block.find_values(dep_date_tag).str(0))
group = block.find_value('_pdbx_deposit_group.group_id')
writer.writerow([code, na, vs, vm, d_min, dep_date, group])
After a few checks (see examples/matthews.py) we may decide to use
only those PDB entries in which VM and VS are consistent.
To make things more interesting we plot separately depositions from before
and after 1/1/2015.
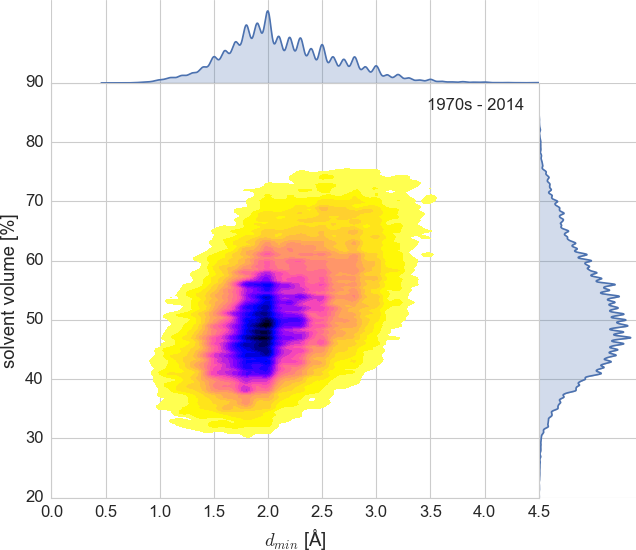
About 90,000 PDB entries (up to 2014)
plotted as intentionally undersmoothed kernel density estimate.
The code used to produce this plot is in examples/matthews.py.¶
Ripples in the right subplot show that in many entries VS is reported as an integer, so we should calculate it ourselves (like Weichenberger & Rupp) for better precision. Ripples in the top plot show that we should use a less arbitrary metric of resolution than dmin (but it’s not so easy). Or we could just smooth them out by changing parameters of this plot.
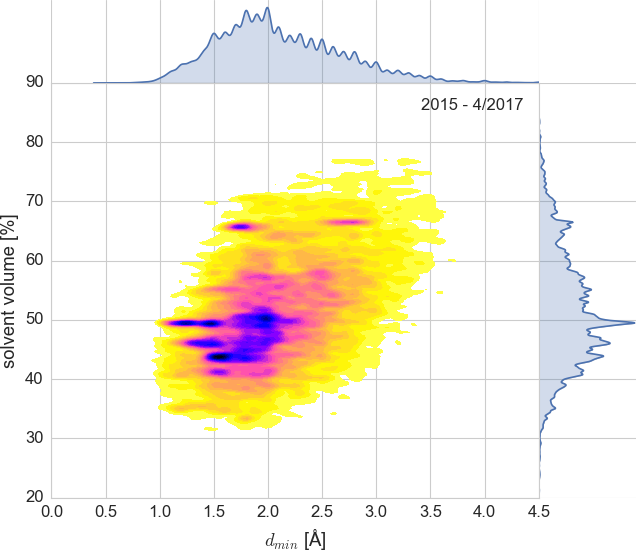
About 17,000 PDB entries (2015 - April 2017)
plotted as intentionally undersmoothed kernel density estimate.
The code used to produce this plot is in examples/matthews.py.¶
On the left side of the yellow egg you can see dark stripes caused by group depositions, which were introduced by the PDB in 2016. They came from two European high-throughput beamlines and illustrate how automated software can analyze hundreds of similar samples (fragment screening) and submit them quickly to the PDB.
We can easily filter out group depositions – either using the group IDs extracted from mmCIF files (we do this for pdb-stats) or, like W&B, by excluding redundant entries based on the unit cell and VM.
Not all the dark spots are group depositions. For example, the one at VS≈66.5%, dmin 2.5–3A is proteasome 20S studied over years by Huber et al, with dozens of PDB submissions.
Weights¶
Let’s say we want to verify consistency of molecular weights. First, let us look at chem_comp tables:
loop_
_chem_comp.id
_chem_comp.type
_chem_comp.mon_nstd_flag
_chem_comp.name
_chem_comp.pdbx_synonyms
_chem_comp.formula
_chem_comp.formula_weight
ALA 'L-peptide linking' y ALANINE ? 'C3 H7 N O2' 89.093
ARG 'L-peptide linking' y ARGININE ? 'C6 H15 N4 O2 1' 175.209
ASN 'L-peptide linking' y ASPARAGINE ? 'C4 H8 N2 O3' 132.118
ASP 'L-peptide linking' y 'ASPARTIC ACID' ? 'C4 H7 N O4' 133.103
CYS 'L-peptide linking' y CYSTEINE ? 'C3 H7 N O2 S' 121.158
EDO non-polymer . 1,2-ETHANEDIOL 'ETHYLENE GLYCOL' 'C2 H6 O2' 62.068
...
We expect that by using molecular weights of elements and simple arithmetic
we can recalculate _chem_comp.formula_weight from _chem_comp.formula.
The full code is in examples/weights.py.
It includes a function that converts 'C2 H6 O2' to {C:2, H:6, O:2}.
Here we only show the lines that sum the element weights
and compare the result:
def check_chem_comp_formula_weight(block):
for cc in block.find('_chem_comp.', ['id', 'formula', 'formula_weight']):
if cc.str(0) in ('UNX', 'UNL'): # unknown residue or ligand
continue
fdict = formula_to_dict(cc.str(1).title())
calc_weight = sum(n * Element(e).weight for (e, n) in fdict.items())
diff = calc_weight - cif.as_number(cc[2])
if not abs(diff) < 0.1: # also true if diff is NaN
print('%s %s %-16s % 9.3f - %9s = %+.3f' %
(block.name, cc[0], cc.str(1), calc_weight, cc[2], diff))
This script prints differences above 0.1 u:
3A1R DOD D2 O 20.028 - 18.015 = +2.013
5AI2 D8U D 1 2.014 - ? = +nan
5AI2 DOD D2 O 20.028 - 18.015 = +2.013
4AR3 D3O O 1 15.999 - 22.042 = -6.043
4AR4 D3O O 1 15.999 - 22.042 = -6.043
4AR5 DOD D2 O 20.028 - 18.015 = +2.013
4AR6 DOD D2 O 20.028 - 18.015 = +2.013
4B1A K3G MO12 O40 P 1 1822.350 - 1822.230 = +0.120
4BVO E43 H2 O40 W12 6 2848.072 - 2848.192 = -0.120
...
5FHW HFW Hf O61 P2 W17 4341.681 - 4343.697 = -2.016
...
The differences are few and minor. We see a few PDB entries with the weight of D2O set to the weight of H2O. The second line shows a missing weight. The differences +0.12 and -0.12 next to Mo12 and W12 probably come from the 0.01u difference in the input masses of the elements. In two entries D3O is missing D in the formula, and -2.016 in HFW suggests two missing hydrogens.
Now let us try to re-calculate _entity.formula_weight from the chem_comp
weights and the sequence.
The PDB software calculates it as the sum of components in the chain,
minus the weight of N-1 waters.
In case of nucleic acids also PO2 is subtracted
(why not PO3? – to be checked).
And in case of microheterogeneity only the main conformer is taken into
account. As the PDB software uses single precision for these computations,
we ignore differences below 0.003%, which we verified to be enough to
account for numerical errors.
def check_entity_formula_weight(block):
# read chem_comp weights from the file, e.g. THR: 119.120
cc_weights = {cc.str(0): cif.as_number(cc[1]) for cc in
block.find('_chem_comp.', ['id', 'formula_weight'])}
# read polymer types from _entity_poly.type
poly_types = {ep.str(0): ep.str(1) for ep in
block.find('_entity_poly.', ['entity_id', 'type'])}
# read and sort sequences from _entity_poly_seq
entity_seq = collections.defaultdict(list)
for m in block.find('_entity_poly_seq.', ['entity_id', 'num', 'mon_id']):
entity_seq[m.str(0)].append((cif.as_int(m[1]), cif.as_string(m[2])))
for seq in entity_seq.values():
seq.sort(key=lambda p: p[0])
# read _entity.formula_weight values
f_weights = block.find('_entity.', ['id', 'formula_weight'])
entity_weights = {ent.str(0): cif.as_number(ent[1]) for ent in f_weights}
# calculate weight from sequences and compare with _entity.formula_weight
for ent, seq in entity_seq.items():
# in case of microheterogeneity take the first conformer
main_seq = [next(g) for _, g in itertools.groupby(seq, lambda p: p[0])]
weight = sum(cc_weights[mon_id] for (num, mon_id) in main_seq)
weight -= (len(main_seq) - 1) * H2O_MASS
if 'ribonucleotide' in poly_types[ent]: # DNA or RNA
weight -= PO2_MASS
diff = weight - entity_weights[ent]
if abs(diff) > max(0.1, 3e-5 * weight):
print('%4s entity_id: %2s %10.2f - %10.2f = %+8.3f' %
(block.name, ent, weight, entity_weights[ent], diff))
Running this on a local copy of the PDB database prints 26 lines, and the difference is always (except for 4PMN) the mass of PO2 showing that we have not fully reproduced the rule when to subtract this group.
...
4D67 entity_id: 44 1625855.77 - 1625917.62 = -61.855
1HZS entity_id: 1 1733.88 - 1796.85 = -62.973
2K4G entity_id: 1 2158.21 - 2221.18 = -62.974
3MBS entity_id: 1 2261.35 - 2324.33 = -62.974
1NR8 entity_id: 2 2883.07 - 2946.05 = -62.974
3OK2 entity_id: 1 3417.14 - 3354.17 = +62.968
...
Disulfide bonds¶
If we’re curious what residues take part in disulfide bonds, we could
write a little script that inspects _struct_conn.
But to show something else, here we use gemmi grep, a little
utility documented in a separate section.
First we try it on a single entry:
$ gemmi grep --delimiter=' ' _struct_conn.conn_type_id \
> -a _struct_conn.ptnr1_label_comp_id -a _struct_conn.ptnr1_label_atom_id \
> -a _struct_conn.ptnr2_label_comp_id -a _struct_conn.ptnr2_label_atom_id \
> 5CBL
5CBL disulf CYS SG BME S2
5CBL covale ILE CD1 BME C2
Then we pipe the output through Unix shell tools.
| grep disulf selects disulfide bonds.
| awk '{ print $3, $4 "\n" $5, $6 }' changes each line into two.
The first output line above becomes:
CYS SG
BME S2
Then we run it on the whole PDB archive, sort, count, and print stats:
$ gemmi grep --delimiter=' ' _struct_conn.conn_type_id \
> -a _struct_conn.ptnr1_label_comp_id -a _struct_conn.ptnr1_label_atom_id \
> -a _struct_conn.ptnr2_label_comp_id -a _struct_conn.ptnr2_label_atom_id \
> /hdd/mmCIF \
> | grep disulf | awk '{ print $3, $4 "\n" $5, $6 }' \
> | sort | uniq -c | sort -nr
367980 CYS SG
274 DCY SG
28 BME S2
23 SO4 S
19 CY3 SG
14 MET SD
13 MTN S1
12 V1A S3
10 MPT SG
8 NCY SG
7 LE1 SG
7 CSX SG
6 CSO SG
5 GSH SG2
5 DTT S1
4 SC2 SG
4 MRG S24
3 H2S S
3 DTT S4
3 1WD S7
2 P8S S1
2 MTE S2'
2 MTE S1'
2 EPE S
2 DHL SG
2 DCD S1
2 CSD SG
2 COM S1
2 6LN S2
2 6LN S1
2 4K3 S4
2 3C7 S01
2 2ON S2
1 SCN S
1 RXR SD
1 RXR S10
1 MEE S
1 G47 SG
1 COA S1P
1 6ML S1
1 5O8 SBH
And what other bond types are annotated in _struct_conn?
$ gemmi grep -O -b _struct_conn.conn_type_id /hdd/mmCIF/ | sort | uniq -c
501851 covale
184231 disulf
4035115 hydrog
1394732 metalc
Chemical Component Dictionary¶
Now let’s look at data from
the components.cif from CCD.
This file describes all monomers (residues,
ligands, solvent molecules) in PDB entries.
As an exercise, let’s check heavy atoms in selenomethionine:
>>> from gemmi import cif
>>> cif.read('components.cif')
<gemmi.cif.Document with ... blocks (000, 001, 002...)>
>>> _.find_block('MSE')
<gemmi.cif.Block MSE>
>>> _.find('_chem_comp_atom.', ['atom_id', 'type_symbol'])
<gemmi.cif.Table 20 x 2>
>>> [':'.join(a) for a in _ if a[1] != 'H']
['N:N', 'CA:C', 'C:C', 'O:O', 'OXT:O', 'CB:C', 'CG:C', 'SE:SE', 'CE:C']
One may wonder what is the heaviest CCD component:
>>> ccd = cif.read('components.cif')
>>> max(ccd, key=lambda b: float(b.find_value('_chem_comp.formula_weight')))
<gemmi.cif.Block WO2>
>>> _.find_value('_chem_comp.formula')
'"O62 P2 W18"'
Or which one has the largest number of heavy atoms:
>>> el_tag = '_chem_comp_atom.type_symbol'
>>> max(ccd, key=lambda b: sum(el != 'H' for el in b.find_values(el_tag)))
<gemmi.cif.Block X12>
>>> _.find_value('_chem_comp.formula')
'"C96 H153 N31 O25"'
The components.cif file is big, so we may want to split it into
multiple files. As an example, here we create a new document with only
one block, and we write it to a file:
>>> block = ccd['X12']
>>> d = cif.Document()
>>> d.add_copied_block(block)
<gemmi.cif.Block X12>
>>> d.write_file('X12.cif')
This example can be expanded to write multiple selected blocks
to a file, as in examples/sub_ccd.py.
To write a single block, we could simply call:
>>> ccd['X12'].write_file('X12.cif')
In the next example we delete things we do not need. Let us write only components on letter A to a new file. Additionally, we remove the descriptor category:
>>> ccd = cif.read('components.cif')
>>> len(ccd)
25219
>>> to_be_deleted = [n for n, block in enumerate(ccd) if block.name[0] != 'A']
>>> for index in reversed(to_be_deleted):
... del ccd[index]
...
>>> len(ccd)
991
>>> for block in ccd:
... block.find_mmcif_category('_pdbx_chem_comp_descriptor.').erase()
...
>>> ccd.write_file('A.cif')
The examples here present low-level, generic handling of CCD as a CIF file. If we would need to look into coordinates, it would be better to use higher level representation of the chemical component, which is provided by gemmi::ChemComp.